
Molecular Dynamics - Sao Carlos Physics Institute (MD-SCPI)
Overview
Study and evaluation of the un/binding kinetics of Mineralocorticoid (MR) and a mutation (MR_S810L) receptor steroid agonist Aldosterone (Aldo), Cortisol (Col), and Progesterone (Str) ligands using Molecular Dynamics (MD) and Monte Carlo (MC) simulations, LiGaMD and LiBELa softwares respectively.
Objectives
- Prepare the input files, PDB and mol files, of the Mineralocorticoid (MR) and a mutation (MR_S810L) proteins and the ligands of interest: Aldosterone (Aldo), Cortisol (Col), and Progesterone (Str)
- Create a Molecular Dynamics (MD) simulation for the MR/MR_S810L-aldo/col/str systems using amber and LiGaMD
- Create a Monte Carlo (MC) simulation for the simple and dimeral MR/MR_S810L-aldo/col/str systems using LiBELA
- Analyze the un/binding events in the MD and MC simulations using PyEMMA, how fast does the ligand scape of the protein?
- Contrast MD and MC simulations, does the MC simulation show the same behavior as MC? can both simulations emulate binding events?
Links of Interest
- PDB-rcsb
- GaMD
- GamD in openMM
- GamD
- OpenMM
- LiBELa
- Amber Manuals
- Chimera User Guide - commands
- Living Journal of Computational Molecular Science
- Tese_Alessandro_Silva_Nascimento.pdf, pag 127
- Highly accurate protein structure prediction with AlphaFold
- Aldosterone and mineralocorticoid receptors: Orphan questions
- Ligand Dissociation from Estrogen Receptor Is Mediated by Receptor Dimerization: Evidence from Molecular Dynamics Simulations
Reports
Reports made during the internship. Here you can find weekly updates and the final report with the results obtained.
Weekly Reports
Final Report
Output Files
Example output files for the MR-AS4 system obtained in the summer internship using python, LiBELa, and LiGaMD.
MR-AS4 MD simulation
MR-AS4 MC simulation
MD Energies
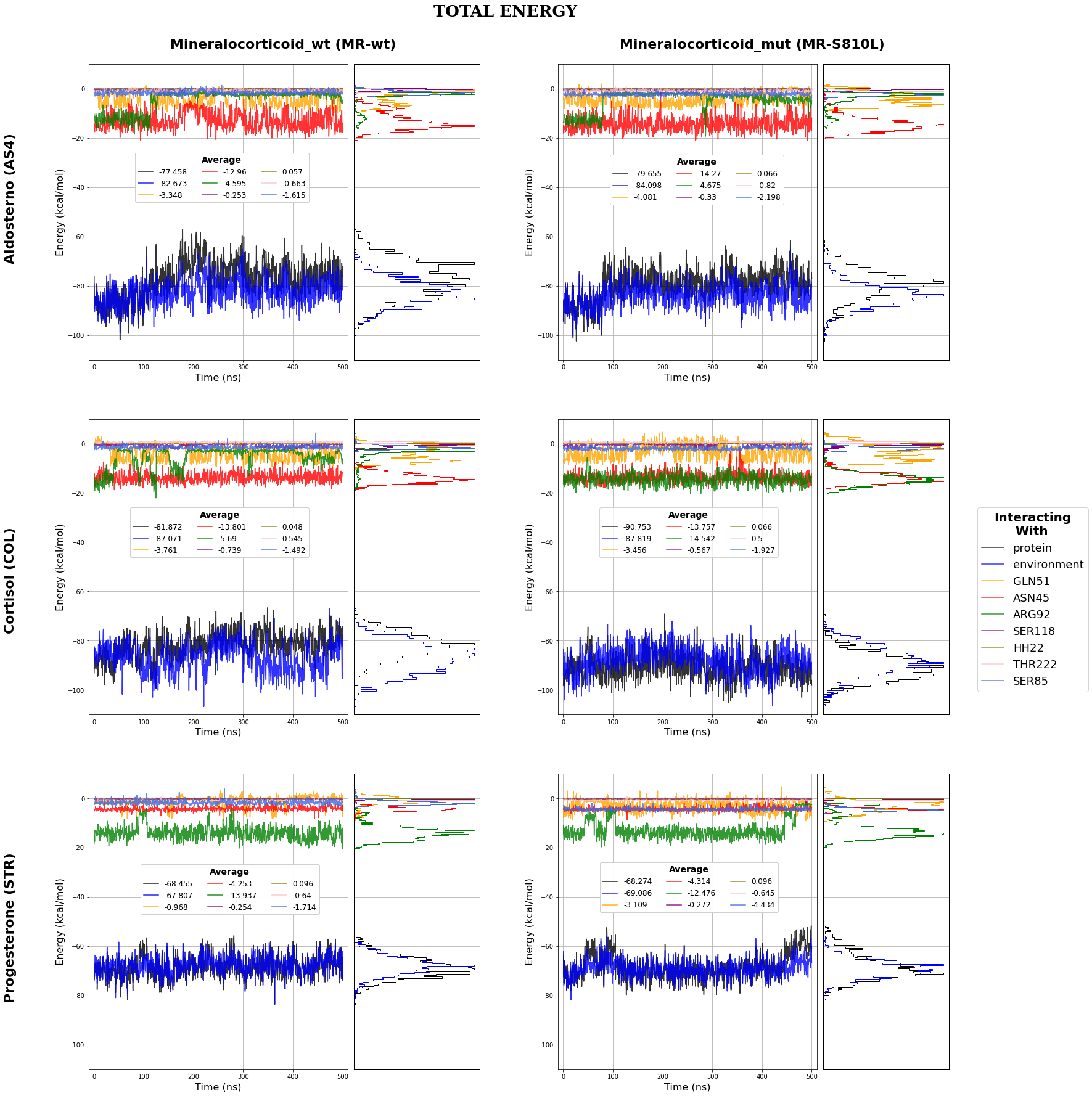
This image is obtained using the PlotEnergies.ipynb notebook that use jscatter pacakge to import dat files and matplotlib to plot.
MC Energies
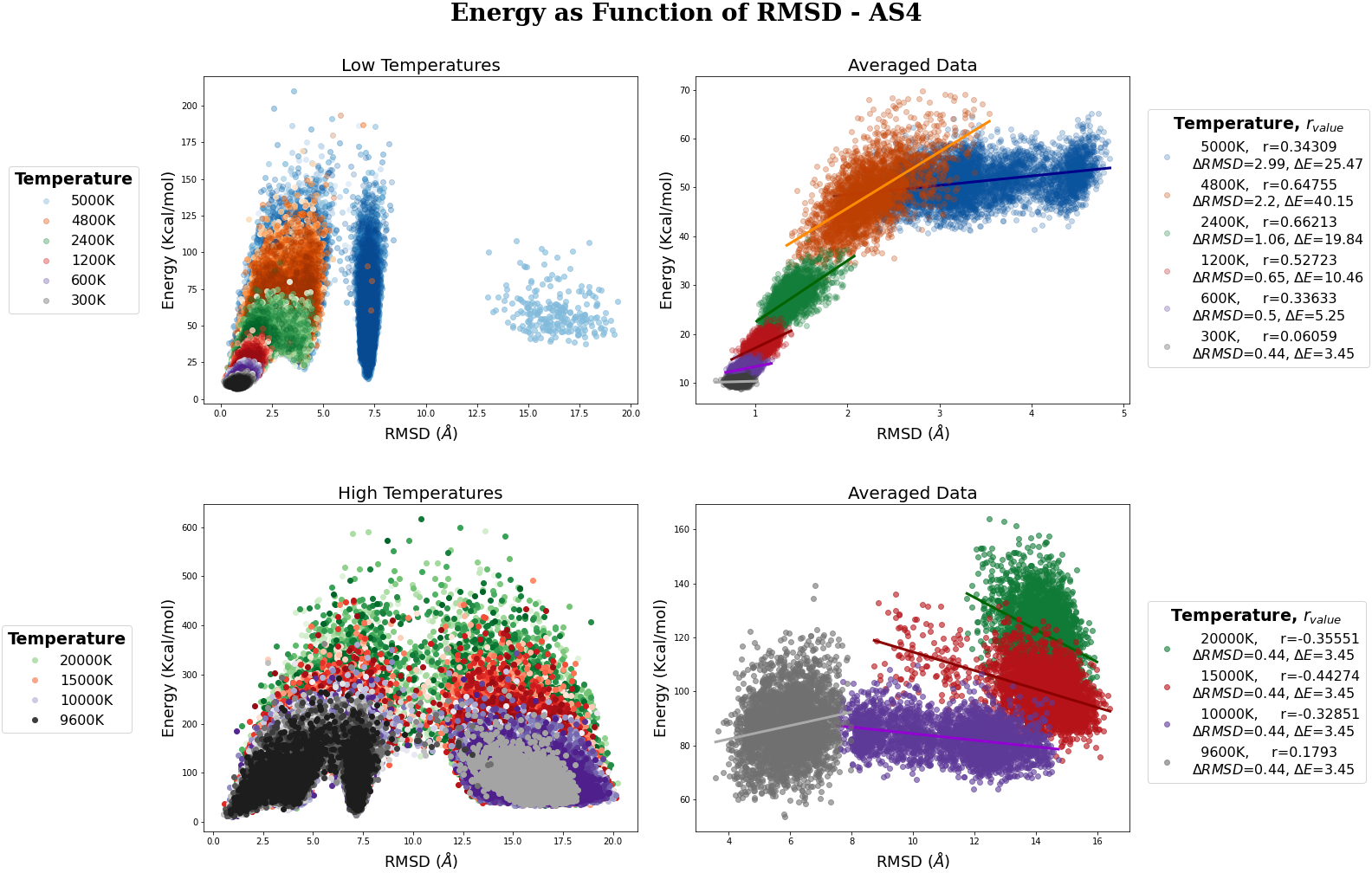
MC RMSD
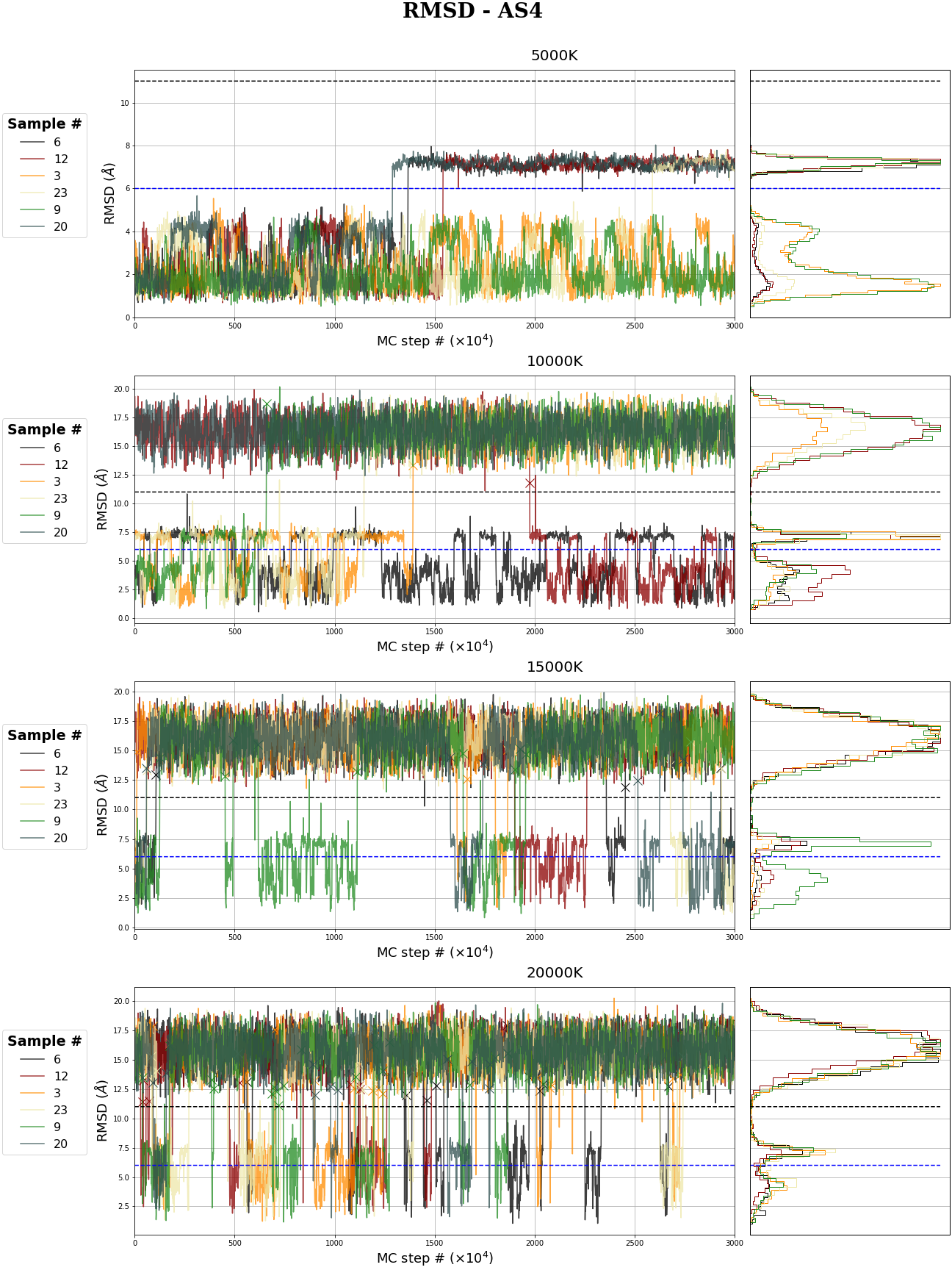
MC Analysis with PyEMMA
States
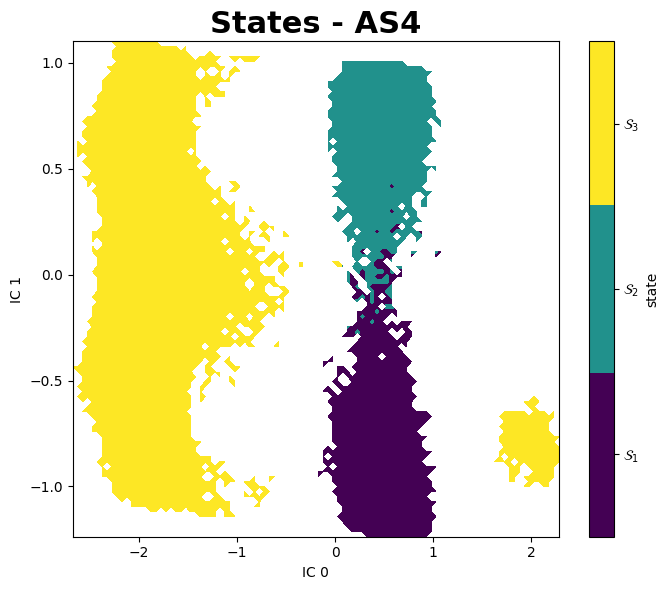
Examples States
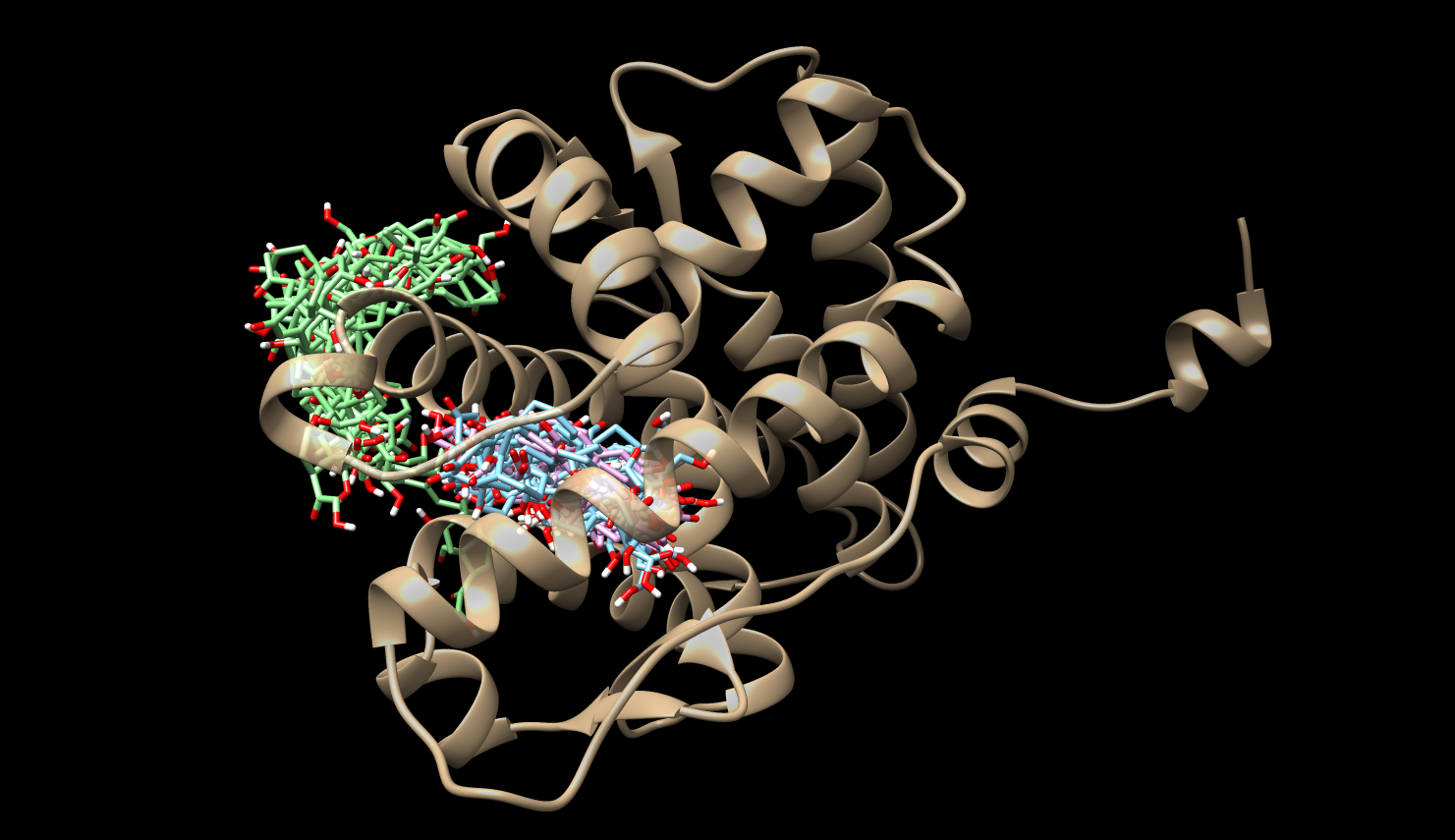
These image are obtained using the notebooks Plots_MC.ipynb, which use jscatter pacakge to import dat files and matplotlib to plot, and PyEMMA_AS4.ipynb, which uses PyEMMA to analyze the simulatiosn outputs.
Mean First Passage Times (MFPTs)
MR-Aldo| 1 | 2 | 3 | |
|---|---|---|---|
| 1 | 0.0 | 1363.7 | 21452.1 |
| 2 | 1417.9 | 0.0 | 21493.2 |
| 3 | 41508.3 | 41498.5 | 0.0 |
| 1 | 2 | 3 | |
|---|---|---|---|
| 1 | 0.0 | 2491.7 | 12451.0 |
| 2 | 3296.1 | 0.0 | 12442.1 |
| 3 | 38690.2 | 37897.6 | 0.0 |
| Ligand | MFPT bound → other | MFPT other → bound |
|---|---|---|
| Aldo | 511.8 ± 8.3 | 34229.1 ± 5028.2 |
| Aldomut | 797.7 ± 16.4 | 38229.3 ± 5514.4 |
Conclusions
- Although LiGaMD-1/2 has demonstrated a tremendous ability to reproduce un/binding process for the systems of interest, MR-MRmut/Aldo-Col-Str, the algorithm does not reproduce un/binding events, no matter how long the simulation is or how high the boost parameters are. This is because the ligand is inside a pocket with a high barrier, the electrical potential between the ligand and protein is very strong and the boost is not enough.
- LiBELa is able to reproduce un/binding in MR-MRmut/Aldo-Col-Str systems, even when it simulates only the dynamics of the ligand and considers the protein as fixed. This shows how powerful the software is even in systems where the binding pocket is buried, which makes dissociation difficult.
- The MC simulations reveal that the mutation of MR (MRS810L) makes the unbinding longer, i.e., for the ligand to escape more LiBELa steps are required. The easiest escaping ligand is the Str, which can be explained because it does not have the same oxygens molecules as the others and make the interaction with the protein weaker; while Col and Aldo need a similar number of LiBELa steps to escape from the MR protein.
- PyEMMA package shows a great solution for analyzing MC simulation since using it in the generated data it is possible to find different states of the system: inside the pocket (bound state), inside the pocket but flipped, and an unbound state (where the ligand is outside the protein); the software even shows the different existing pathways for the ligand to escape by classify them into two different microstates.
References
Proteins and Ligands
[1] Chantal Hellal-Levy, Jérôme Fagart, Anny Souque, and Marie-Edith Rafestin-Oblin. Mechanistic aspects of mineralocorticoid receptor activation. Kidney International, 57(4):1250–1255, 2000.
[2] Marie-Edith Rafestin-Oblin, Anny Souque, Brigitte Bocchi, Gregory Pinon, Jerome Fagart, and Alain Vandewalle. The Severe Form of Hypertension Caused by the Activating S810L Mutation in the Mineralocorticoid Receptor Is Cortisone related. Endocrinology, 144(2):528–533, 02 2003.
[3] David S. Geller, Anita Farhi, Nikki Pinkerton, Michael Fradley, Michael Moritz, Adrian Spitzer, Gretchen Meinke, Francis T. F. Tsai, Paul B. Sigler, and Richard P. Lifton. Activating mineralocorticoid receptor mutation in hypertension exacerbated by pregnancy. Science, 289(5476):119–123, 2000.
[4] Jérôme Fagart, Jessica Huyet, Grégory M. Pinon, Marina Rochel, Claudine Mayer, and Marie-Edith Rafestin-Oblin. Crystal structure of a mutant mineralocorticoid receptor responsible for hypertension. Nature Structural & Molecular Biology, 12(6):554–555, Jun 2005.
[5] John W. Funder. Aldosterone and mineralocorticoid receptors: Orphan questions. Kidney International, 57(4):1358–1363, 2000.
Software
[6] K. Belfon I.Y. Ben-Shalom J.T. Berryman S.R. Brozell D.S. Cerutti T.E. Cheatham III G.A. Cisneros V.W.D. Cruzeiro T.A. Darden R.E. Duke G. Giambasu M.K. Gilson H. Gohlke A.W. Goetz R. Harris S. Izadi S.A. Izmailov K. Kasavajhala M.C. Kaymak E. King A. Ko-valenko T. Kurtzman T.S. Lee S. LeGrand P. Li C. Lin J. Liu T. Luchko R. Luo M. Machado V. Man M. Manathunga K.M. Merz Y. Miao O. Mikhailovskii G. Monard H. Nguyen K.A. O’Hearn A. Onufriev F. Pan S. Pantano R. Qi A. Rahnamoun D.R. Roe A. Roitberg C. Sagui S. Schott-Verdugo A. Shajan J. Shen C.L. Simmerling N.R. Skrynnikov J. Smith J. Swails R.C. Walker J. Wang J. Wang H. Wei R.M. Wolf X. Wu Y. Xiong Y. Xue D.M. York S. Zhao D.A. Case, H.M. Aktulga and P.A. Kollman (2022). Amber 2022. University of California, San Francisco.
[7] Jinan Wang and Yinglong Miao. Ligand gaussian accelerated molecular dynamics 2 (ligamd2): Improved calculations of ligand binding thermodynamics and kinetics with closed protein pocket. Journal of Chemical Theory and Computation, 19(3):733–745, 2023. PMID: 36706316.
[8] Yinglong Miao, Apurba Bhattarai, and Jinan Wang. Ligand gaussian accelerated molecular dynamics (ligamd): Characterization of ligand binding thermodynamics and kinetics. Journal of Chemical Theory and Computation, 16(9):5526–5547, 2020. PMID: 32692556.
[9] Yinglong Miao, Apurba Bhattarai, and Jinan Wang. Ligand gaussian accelerated molecular dynamics (ligamd): Characterization of ligand binding thermodynamics and kinetics. bioRxiv, 2020.
[10] Heloisa dos Santos Muniz and Alessandro S. Nascimento. Ligand- and receptor-based docking with LiBELa. Journal of Computer-Aided Molecular Design, 29(8):713–723, Aug 2015.
[11] Pablo R. Arantes, Marcelo D. Polêto, Conrado Pedebos, and Rodrigo Ligabue-Braun. Making-it-rain: Cloud-based molecular simulations for everyone, August 2021.
[12] Matthew M. Copeland, Hung N. Do, Lane Votapka, Keya Joshi, Jinan Wang, Rommie E. Amaro, and Yinglong Miao. Gaussian Accelerated Molecular Dynamics in OpenMM. The Journal of Physical Chemistry B, 126(31):5810–5820, 2022. PMID: 35895977.
[13] Jinan Wang, Pablo R. Arantes, Apurba Bhattarai, Rohaine V. Hsu, Shristi Pawnikar, Yu-ming M. Huang, Giulia Palermo, and Yinglong Miao. Gaussian accelerated molecular dynamics: Principles and applications. WIREs Computational Molecular Science, 11(5):e1521, 2021.
[14] Geraldo Rodrigues Sartori and Alessandro S. Nascimento. Comparative Analysis of Electrostatic Models for Ligand Docking. Frontiers in Molecular Biosciences, 6, 2019.
[15] Heloisa S. Muniz and Alessandro S. Nascimento. Towards a critical evaluation of an empirical and volume-based solvation function for ligand docking. PLOS ONE, 12(3):1–19, 03 2017.
Theory
[16] Tamar Schlick. Molecular Modeling and Simulation: An Interdisciplinary Guide, volume 21 of Interdis- ciplinary Applied Mathematics. Springer New York, NY, Berlin, Heidelberg, 2 edition, August 2010.
[17] A.R. Leach. Molecular Modelling: Principles and Applications. Prentice Hall, 2 edition, 2001.